Searching for Control of the Bacillus Subtilis Spore Surface
{kind=link}
To most people, protein is just a necessary part of our diets. But to biologists, proteins are everything. Proteins make up around 15 percent of our body mass [1] and are involved in virtually every process necessary to keep us alive. From digesting our food to repairing our bodies, proteins are hard at work [2].

Charlotte Thomas
Proteins are able to take on such a wide variety of roles because of their ability to fold into an endless number of different configurations, which in turn confer different functions. The endless number of protein functions makes them an incredibly important bioengineering tool, but their utility is unfortunately limited by their fragility. When taken out of the environment the protein evolved to function in, known as its native environment, the protein's intricate folded shape often falls apart, which causes the protein to lose its function [2]. If proteins could consistently function efficiently outside of their native environment, this would vastly improve many bioengineering processes, such as making vaccines and removing dangerous metals from the environment [3].
Over the years, scientists have been looking to create methods to make this happen. One current method involves the use of the bacterial spores of Bacillus subtilis, or B. subtilis. The spores of B. subtilis have protein shells on the surface, called spore coat, which allows them to function robustly even under the harshest conditions. My research project, which began in fall 2021 in the lab of Kang Wu, an associate professor of chemical engineering at the University of New Hampshire, and continued through the summer of 2022 as part of the Research Experience and Apprenticeship Program (REAP) at UNH, would come to focus on improving this method.
All About Spores
B. subtilis is a common kind of rod-shaped bacteria, often found in the soil. It is an extremely well-studied bacteria, often used in industrial processes to mass-produce enzymes [4]. What makes this bacterium special is its ability to sporulate. Sporulation is a survival mechanism for bacteria, initiated under starvation conditions. Sporulation involves the bacteria forming a protective outer shell and entering into a metabolically dormant state. (Figure 1.) This allows the bacteria to survive until conditions improve. What makes this ability relevant to utilizing proteins for bioengineering purposes is that due to the robust nature of the spore, foreign proteins displayed on its outer shell can be functional in conditions far different from their native environment [5].
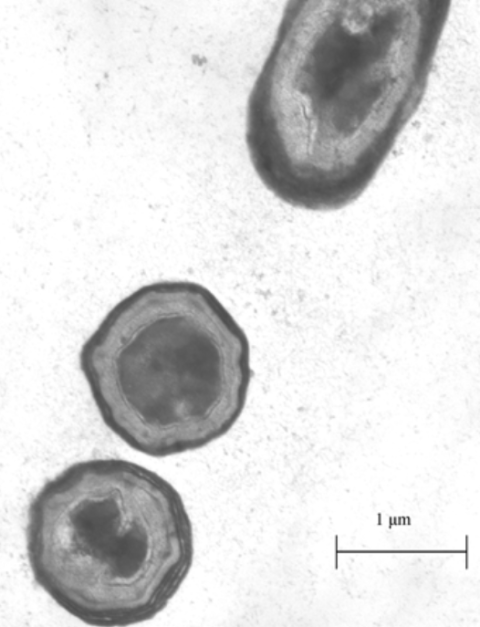
Figure 1: Image of B. subtilis spores under a microscope [7].
Dr. Wu’s lab studies the display of proteins and peptides on the surface of bacterial spores. The amount of protein on the spore surface is relevant in many processes, and overproduction or underproduction of the spore coat proteins can lead to suboptimal results [3]. The production level of a protein is controlled by the expression level of the gene encoding that protein. In turn, the expression level of a gene is controlled by a genetic element called a promoter. Promoters can turn gene expression on or off by interacting with a specific chemical called an inducer. Using promoters with specific inducer chemicals, scientists can engineer inducible promoter systems which allow them to control when genes are turned on. Currently, an inducible promoter system constructed using the natural promoter of one of the B. subtilis spore coat proteins has not been characterized [3, 6].
In Dr. Wu’s lab, I was given the job of continuing the work of a project started by a previous student. The goal of this project was to genetically engineer B. subtilis so that the amount of a certain protein on the spore surface could be controlled by the scientist, by constructing and characterizing an inducible promoter for spore coat protein expression. This goal is important because characterizing promoters allows scientists to have more tools available for bioengineering.
The Research Plan
Extensive lab work was needed to accomplish my research objective. There were three main steps of the research plan: constructing plasmids, integrating them into the B. subtilis genome, and inducing sporulation.

Figure 2: Illustration of the plasmid being constructed in this experiment. The backbone plasmid was cut with restriction enzymes in order to incorporate a fragment of DNA containing an inducible promoter for a spore coat protein gene linked to a GFP gene. Click to enlarge.
The first step of the research plan was to construct plasmids. (Figure 2.) Plasmids are circular pieces of DNA commonly used as a tool for genetic engineering. This is done by incorporating a gene of interest into the plasmid’s DNA. In this case, we wanted to create plasmids containing the inducible promoter sequences. Constructing a new plasmid has five general steps. First, you must make many copies of your gene of interest, and of the plasmid to which you want to add your gene of interest, known as the backbone plasmid. Second, you cut the DNA of the gene of interest and backbone plasmid by adding restriction enzymes, which create sticky ends where the two pieces of DNA can be linked together. Third, the sticky ends of the gene of interest and backbone plasmid are glued together using an enzyme called DNA ligase. Fourth, these plasmids are put into a host bacterium, usually E. coli, through a procedure called a transformation. Fifth, bacteria that successfully take up the plasmid are further cultured, and then the plasmid is extracted.
After constructing these plasmids, the second step of the research plan was to integrate them into the B. subtilis genome. This involves an arduous day-long procedure where you cut the plasmid DNA and mix it with B. subtilis in a chemical cocktail that weakens the bacteria and allows the plasmid DNA to enter the cell. The plasmid DNA then goes through a recombination event where a subset of its DNA sequence is copied over into the B. subtilis genome.
The final step in our research plan was to induce sporulation within the gene edited B. subtilis and observe the results. The sporulation procedure involves putting the transgenic B. subtilis into a special nutrient-deficient media and shaking the
Figure 3: Illustration of a B. subtilis spore, demonstrating the outer spore coat surrounding the spore core containing the bacterial DNA. It also depicts spore coat proteins attached to green fluorescent proteins being presented on the outer spore coat. Click to enlarge.
mixture in an incubator for three to seven days. The lack of some key nutrients in the media more quickly activates the pathways for sporulation.
Prior to my work on the project, designs for a control plasmid and a plasmid containing the inducible promoter were developed by Dr. Wu. The control plasmid contained the natural spore surface protein sequence and promoter sequence. The inducible promoter plasmid contained the same, except before the protein sequence, there was a LacI binding site sequence. The LacI binding sequence is what would make the promoter inducible, by providing a place for the LacI regulator protein to bind in absence of the inducer molecule, and therefore control expression of the spore surface protein gene. When the inducer molecule is present, the gene can be expressed; if it is absent, the gene will not be expressed. The LacI inducer molecule used in this experiment is isopropyl ß-D-1-thiogalactopyranoside, typically referred to as IPTG.
Connected to the gene for the spore surface protein in both plasmids was the gene for the green fluorescent protein or GFP. (Figure 3.) This protein has the unique function of being able to glow green under UV light, which allows for easy testing of the functionality of the promoters.
If the design worked correctly, the control strain should have been able to glow green without our inducer chemical (IPTG) and the inducible promoter strain should have only glowed green if the inducer chemical was present. Whether or not a sporulation sample was glowing green could be determined by a machine called a microplate reader, which can quickly quantify the amount of fluorescence being emitted from a source.
Navigating Unexpected Results
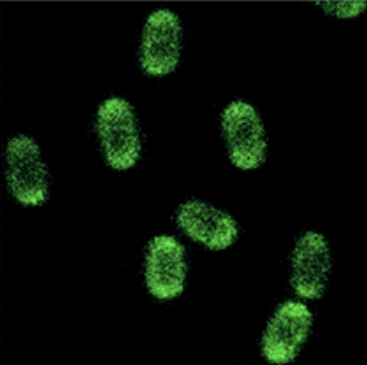
Figure 4: This image shows how B. subtilis spores should have appeared when presenting GFP on their surface under a fluorescent microscope [8].
I was able to complete the first part of my research plan, the construction of the plasmids, before the start of the summer. Having spent a year on this first step I was excited to finally be able to move on. I was able to complete step two, integrating the plasmids into the B. subtilis genome within a week. Finally, I reached the last step of my research project: analyzing the sporulation results. I set up my flasks full of sporulation medium and transgenic B. subtilis and left them to shake. One week later, the medium had turned a dark yellow, and the spores were ready for analysis. After nearly a year of work at this point, I was more than ready to see some results.
I cleaned off my spores, and input them to the microplate reader, expecting to see the previously described pattern of fluorescence up on the microplate reader screen. What came up greatly surprised me. None of the spore samples were fluorescing. All the samples showed the same amount of fluorescence as the control spores with no GFP. I had suspected that there could be issues with the fluorescence in the inducible promoters, but the lack of fluorescence even in the control spores, which were supposed to fluoresce no matter the conditions, was very unexpected.
These results meant it was time to step back and reevaluate. The big question I needed to answer was: Why exactly were my spores not glowing? There were many possible answers to this question, but to figure out the culprit, I would have to interrogate the primary suspects.
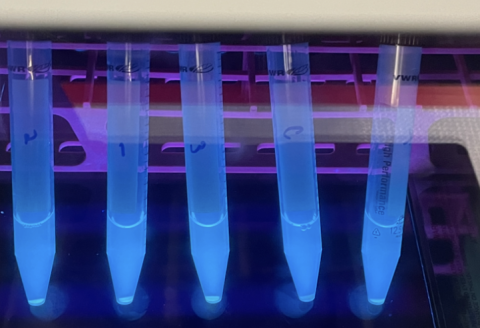
Figure 5: This image depicts test tubes containing E. coli expressing GFP under a fluorescent light allowing its green color to be observed. These test tubes were created to confirm the GFP gene being used was still functional, as part of the troubleshooting process.
First I started with the fluorescence itself. I wondered if there was a problem with the GFP molecule, such as if it had potentially mutated and that was why there was no fluorescence. To test this, I cultured the original bacteria which held the gene for GFP and checked their fluorescence under a UV light. These bacteria were glowing perfectly. So I moved on to the next suspect: the microplate reader. Maybe I had made some sort of user error. Sure enough, when I input the GFP bacteria that glowed bright green under the UV light into the microplate reader, no fluorescence was registered. I adjusted the wavelengths of the microplate reader, and a stark difference between the GFP bacteria and normal bacteria could be seen on screen. Had I found the issue? I retested my spores with these verified machine settings, and still I saw no fluorescence.
For weeks I continued, creating mini experiments to try to figure out why my spores were not glowing. Eventually, my troubleshooting experiments led me to the conclusion that there must be something wrong with the plasmids themselves. This meant I was going to have to restart my project from scratch. After thinking I was just on the cusp of completion, I was now back at step one. But it was through having to restart, that I would come to learn the most.
Beginning, Again
This time around I was determined to make my spores glow. To do this, I knew my plasmid design was going to need to be meticulous. I decided to start with fixing my control plasmid, as the inducible promoter was derived from the control. The one problem for me was that my knowledge of plasmid design was rather limited. So, if I was going to figure out how to make a working plasmid, I had a lot to learn. I spent a few weeks reading all about plasmids and genetics. I pieced together and annotated all parts of the plasmid, in order to try to find the issue. Finally, I experienced one of the highs of my project, when one day I was combing through the plasmid sequence, trying to find the issue, when I spotted something. In the plasmid cut site right before the inducible promoter gene, there was a premature start codon, the DNA sequence which initiates transcription. Because there was a start codon in the cut site, rather than just at the beginning of the gene where it was supposed to be, it was throwing off the translation of the GFP protein, and consequently wrecking it. Finding this issue was very exciting, as it was like finding a needle in a haystack, and now my plasmid redesign had a much better chance of being successful.
With my plasmid redesign finished to the best of my ability, it was time to get back into the lab work. A key thing I learned from my project is that one of the most challenging parts of biological research is the lab work itself. You would think biological lab work would be a straightforward list of procedures. In reality, it never works out according to plan. If a plasmid construction was completed in one go, it would take about two days of work. Even though I had been practicing my lab skills all year, completing my new plasmid construction took over a month. The difficulty comes from the fact that everything builds on the last step, so even one tiny error can result in having to redo several weeks’ worth of work. Problems would arise from the most inane mistakes, such as using a slightly too small DNA collection column, or not making sure the outside of the PCR tube was completely dry, and suddenly all my progress was gone and I was back to square one. Through my experience in the lab, I learned that I had to pay attention to every single detail in order to have success. And when even that didn't work, I learned I had to be patient and keep trying through discouraging patches of time.
Closing Out the Summer
As the summer was coming to a close, my new plasmid construction was finally completed. But time was running out. It took me a bit longer this time, two weeks, to integrate and sporulate my bacteria. I attempted to test for the presence of GFP in my new improved transgenic spores, but time and time again the results were inconclusive, due to a variety of experimental issues. Extensive troubleshooting was needed. Unfortunately, I no longer had the time, as I had begun a different research opportunity and needed to move on.
I am disappointed I didn't accomplish my larger research objective of creating inducible promoters for spore coat protein expression. But I don't think that was the most important outcome of the research. What was important was all I learned. Through this experience, I learned how to successfully perform a variety of wet lab procedures, as well as how to effectively create and execute a research plan, even through difficult periods of troubleshooting. Additionally, this experience allowed me to gain a much more dynamic and nuanced understanding of biology that cannot be gained from lectures alone.
My work on this project stands as the first road mark on the way to my goal of becoming a scientist, where I have begun to learn the fundamentals of research work. I am excited to see how all I have learned from this will advance my scientific work going forward.
I would like to extend my gratitude to Dr. Wu and graduate student Guo Wu for all their guidance, teaching, and help on this project. I would also like to thank the Hamel Center for Undergraduate Research, especially Mr. Dana Hamel and Mr. Nicholas Bencivenga, for gifting me the funding that allowed me to put such extensive time into this work, which in turn allowed me to learn all that I did.
Works Cited
- ZiMian Wang, Wei Shen, Donald P Kotler, Stanley Heshka, Lucian Wielopolski, John F Aloia, Miriam E Nelson, Richard N Pierson, Jr, Steven B Heymsfield. Total body protein: a new cellular level mass and distribution prediction model, The American Journal of Clinical Nutrition, Volume 78, Issue 5, November 2003, Pages 979–984, https://doi.org/10.1093/ajcn/78.5.979
- Koshland, D. E. and Haurowitz, Felix. Protein. Encyclopedia Britannica. 2023, January 26. https://www.britannica.com/science/protein
- Guoyan Z, Yingfeng A, Zabed H, Qi G, Yang M, Jiao Y, Li W, Wenjing S, Xianghui Q. Bacillus subtilis Spore Surface Display Technology: A Review of Its Development and Applications. J Microbiol Biotechnol. 2019 Feb 28;29(2):179-190. doi: 10.4014/jmb.1807.06066. PMID: 30602268.
- Su, Y., Liu, C., Fang, H. et al. Bacillus subtilis: a universal cell factory for industry, agriculture, biomaterials and medicine. Microb Cell Fact 19, 173. 2020. https://doi.org/10.1186/s12934-020-01436-8
- McKenney, P., Driks, A. & Eichenberger, P. The Bacillus subtilis endospore: assembly and functions of the multilayered coat. Nat Rev Microbiol 11, 33–44. 2013. https://doi.org/10.1038/nrmicro2921
- Quynh Anh Nguyen, Wolfgang Schumann, Use of IPTG-inducible promoters for anchoring recombinant proteins on the Bacillus subtilis spore surface, Protein Expression and Purification, Volume 95, 2014, Pages 67-76, ISSN 1046-5928, https://doi.org/10.1016/j.pep.2013.11.018.https://www.sciencedirect.com/science/article/pii/S1046592813002623
- Black, E.P., Setlow, P., Hocking, A.D., Stewart, C.M., Kelly, A.L. and Hoover, D.G. Response of Spores to High-Pressure Processing. Comprehensive Reviews in Food Science and Food Safety. 2007. 6: 103-119. https://doi.org/10.1111/j.1541- 4337.2007.00021.x
- Quijano, Juan F., Sahin, Ozgur. Genetically Intact Bioengineered Spores of Bacillus subtilis. ACS Synthetic Biology. 2021. 10 (4), 778-785 DOI: 10.1021/acssynbio.0c00578
Author and Mentor Bios
A Stratham, New Hampshire, native, Charlotte Thomas is a second-year bioengineering student at the University of New Hampshire, where she is also part of the University Honors Program. Through the Research Experience and Apprenticeship Program (REAP), funded by the Hamel Center for Undergraduate Research, Charlotte gained a deep understanding of genetics. Her research project aimed to improve methods of utilizing proteins outside of their native environment using the bacteria Bacillus subtilis. Working on this project helped Charlotte to develop a strong grasp of the problem solving and persistence required for genetic research. After graduation she plans to pursue a Ph.D., studying the biology of aging, with the goal of becoming a professor in that area.
Kang Wu is an associate professor of chemical engineering. She began working at UNH in 2012 and teaches courses in both chemical engineering and bioengineering. Dr. Wu’s research focuses on the synthesis and display of proteins and peptides on bacterial spore surfaces. This has applications ranging from drug delivery, biosensors, bioremediation, and bioprocessing. Dr. Wu mentored Charlotte during her Research Experience and Apprenticeship Program (REAP) project and describes her as a passionate and self-motivated researcher. Dr. Wu has worked with over twenty undergraduate researchers in her lab, many of whom have received grants from the Hamel Center for Undergraduate Research.
Copyright 2023, Charlotte Thomas