












































UNH researchers have advanced our understanding of the mutations that result in color blindness and certain retinal degenerative diseases that cause blindness as a result of their unprecedented look at the structure and function of a protein central to color vision in humans.
Predicting Eye Disease
New National Eye Institute grant supports research into degenerative eye diseases
Tuesday, December 6, 2022
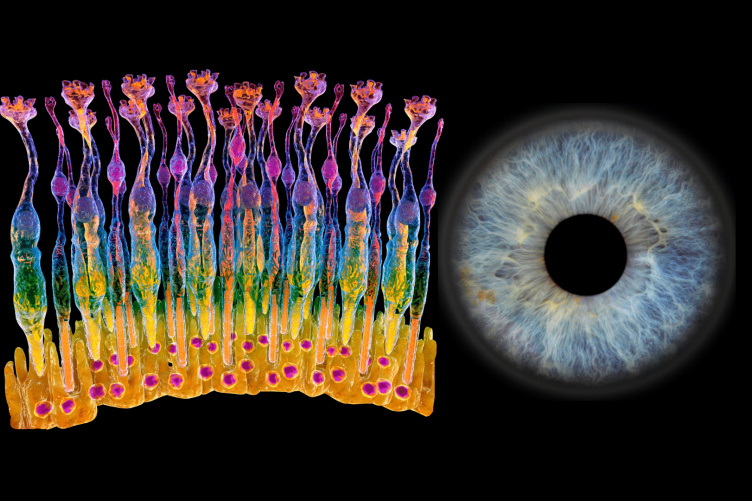
RICK COTE'S RESEARCH EXAMINES DISEASES THAT AFFECT THE PHOTORECEPTOR CELLS IN THE RETINA (LEFT). THE ILLUSTRATION OF THE PHOTORECEPTOR CELLS SHOWS THE SLENDER, ORANGE-TIPPED ROD CELLS AND BULBOUS, YELLOW-TIPPED CONE CELLS THAT ABSORB LIGHT AND TRANSMIT SIGNALS TO THE PHOTORECEPTOR SYNAPSES (TOP OF DIAGRAM).
A recently awarded $1.5 million grant from the U.S. National Institutes of Health’s (NIH) National Eye Institute will support ongoing research into retinal degenerative diseases led by Rick Cote, a professor in the molecular, cellular and biomedical sciences department and the director of the Center of Integrated Biomedical and Bioengineering Research (CIBBR) at UNH. This latest grant extends three decades of continuous funding from NIH to support Cote’s research program examining the visual signaling pathways in rod and cone photoreceptor cells that are responsible for the very first events in the vision process.
Cote’s lab has long studied the visual signaling pathway’s central enzyme, called phosphodiesterase-6 (PDE6), including how this enzyme is activated by light and subsequently how it returns to its dark-adapted state. The current grant supports research into determining the atomic-level structure of PDE6, as well as the activating and inactivating proteins that bind to PDE6 during visual signaling. This structural knowledge of PDE6 and its binding partners is critical for improving our ability to predict whether genetic mutations of PDE6 and/or the proteins that regulate PDE6 are likely to cause visual disorders or blindness.

“Each of us has a multitude of mutations in our genes, and many of these mutations are silent or benign,” described Cote. “We’re trying to predict which mutations in PDE6 are pathogenic and prevent the visual signaling pathway from responding properly to light, ultimately leading to rod photoreceptor cell death and the progressive degeneration of the entire retina.”
"We’re trying to predict which mutations in PDE6 are pathogenic and prevent the visual signaling pathway from responding properly to light, ultimately leading to rod photoreceptor cell death and the progressive degeneration of the entire retina."
Cote’s current research focuses on understanding the molecular defects that cause retinitis pigmentosa, an inherited retinal degenerative disease that typically presents symptoms that begin in childhood or early adulthood and can result in total blindness. Approximately 1 in 3,500 people in the United States have the disease.
“One of the things that distinguishes retinitis pigmentosa from other blinding diseases is that the onset of symptoms occurs in late childhood or adolescence, and are not apparent at birth,” said Cote. “The vision of a person with retinitis pigmentosa begins to deteriorate as they age, starting with night blindness (an inability to see at night), a gradual loss of peripheral vision and eventually total or near-total blindness.”
With the advent of inexpensive DNA sequencing technologies, people at risk of inheriting retinitis pigmentosa or related eye diseases—or children starting to display loss of visual function—can have their genome sequenced to determine whether they harbor pathogenic mutations in the rod and cone PDE6 genes.
“Genetic screening of at-risk individuals will allow us to intervene early on with treatments–before their vision deteriorates — and hopefully prevent the degenerative process,” said Cote.
Therapeutic treatments may take the form of administering drugs that can preserve photoreceptor function or use gene editing to permanently correct the mutation. By mapping known PDE6 mutations to the enzyme’s three-dimensional structure, Cote’s lab will improve our ability to distinguish pathogenic from harmless mutations and give ophthalmologists greater precision in diagnosing and treating retinal diseases.
You can learn more about Cote’s research on his lab website.
This research is funded by the National Eye Institute (NIH).
-
Written By:
Nicholas Gosling '06 | COLSA/NH Agricultural Experiment Station | nicholas.gosling@unh.edu


